1細胞解析は、生命科学研究にとってますます重要になりつつあります。
そこから得られたデータは、無料の統計ソフトRと、バイオ関連のツールが揃ったBioconductorを使って行われます。
この記事では、1細胞解析に使われるBioconductorパッケージについて紹介します。
この記事の内容
1細胞解析の基本の流れ
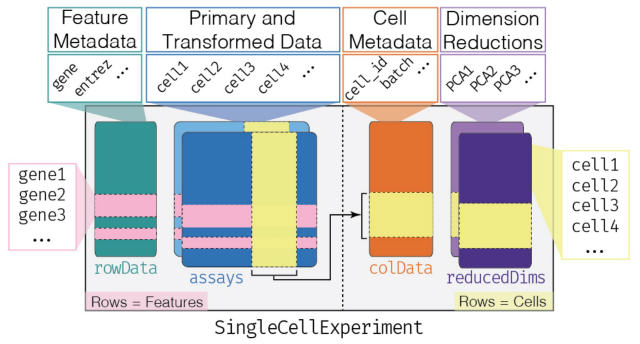
1細胞解析をRで行う上で避けて通れないのがSingleCellExperimentクラスです。
データ構造はこのようになっています。
https://osca.bioconductor.org/data-infrastructure.html
Assaysというテーブルでは、行が遺伝子、列が各細胞を表しています。
そして遺伝子についての情報はrowDataに、細胞についての情報はcolDataに置かれて、これらがつながっているというのがSingleCellExperimentクラスの基本構造です。
このデータは、1細胞解析結果のリードをマッピングさせ、遺伝子ごとの数を数えることで得られます。
また、これを使ってさまざまな解析が行われます。
どのようなRのパッケージがあるのか、代表的なものを紹介します。
データ処理
前処理
1細胞RNA-seqでの前処理は、リードをマッピングさせて定量し、発現値の細胞および遺伝子ごとのカウントマトリックスを得ます。
いろいろなツールがありますが、scPipeやscruffといったBioconductorパッケージもあります。
また、DropletUtilsやtximetaを使えば、Cell RangerやKallisto-Bustoolsといった様々なコマンドラインツールの結果をRにインポートすることができます。
これらのワークフローでは、カウントマトリックスをインポートし、最終的SingleCellExperimentオブジェクトが作成されます。
品質チェック
サンプル調製中の細胞へのダメージやライブラリ調製の失敗などの結果、低品質なライブラリになってしまうこともありえます。
しかしそれでもデータ解析上は「細胞」として扱われてしまうので、ここで除く必要があります。
発現遺伝子が少ないとか、ミトコンドリアのリードが多いというものを除きます。
ドロップレットが必ずしも1細胞を含んでいるとは限らないのでこれらも除く必要があります。
DropletUtilsパッケージでは、観察された各液滴バーコードの頻度などから計算して、細胞を含んでいないドロップレットを区別することが可能です。
1つではなく複数の細胞が同じドロップレットの中に入っていると考えられる場合には、2つはいっていた場合のドロップレットの遺伝子発現量シミュレーションと比較して、1つなのかたくさん入っているかを識別できるscranやscdsといったパッケージで除くことが可能です。
このようにして1細胞のみが入っているドロップレットを選択した後、ダメージを受けている可能性が高い細胞や、そもそもリードが少ない細胞を除外します。
各細胞でのすべての遺伝子カウントの合計として定義されるライブラリーサイズが小さいものや、あるいは発現遺伝子数が非常に少ない細胞、ミトコンドリアゲノムへマップされるリードが多い細胞を除外することが多いです。
最後のミトコンドリアについては、細胞がダメージを受けて細胞膜に穴が開くと、通常の転写産物は小さいので細胞外に抜けていくが、ミトコンドリア転写産物は大きいので細胞外に行くことができず、結果としてミトコンドリア由来のリードの割合が多い細胞はダメージを受けた細胞だという考えに基づいています。
Bioconductorではscaterパッケージでこれらの指標を簡単に計算することができます。
正規化と推定
細胞によってcDNAやPCR増幅効率が違うので、ライブラリー調製でさまざまな誤差が入ってしまいます。これらのノイズを制御する方法もいくつか提案されています。
最も単純な戦略としては、細胞ごとのライブラリーサイズをscaterなどのツールで正規化するということです。
また、1細胞データは多くの遺伝子が0としてカウントされるスパースなデータですが、これは本当に0なのか、それともたまたま読めなかっただけなのかが難しいところです。
これを推定して補正するアルゴリズムなどもいろいろ提案されていますが、これについては長くなるので機会があれば別の記事でまとめようと思います。
遺伝子選択
クラスターリングや次元圧縮に使う遺伝子を選択します。ここでは、ランダムノイズを多く含む遺伝子を除去しながら、有用な情報をもつ遺伝子を拾い上げるというのが目的です。
生命科学・医学の観点から本当に意味のある遺伝子であれば、条件Aと条件Bを比べたときに大きく変動しているはずであり、一方で単なるノイズを見ているだけの遺伝子は条件AでもBでも違いはそれほどないはずです。
そのため、発現量の分散が大きい遺伝子を選択するというのが最も簡単なアプローチということになります。
実際にはもっと複雑で、遺伝子発現量によっても分散は変わりうるので、統計学者の方々がいろいろ工夫した方法を考案しています。
生命医学の研究をする私たちは、統計専門家の方々が作ったアルゴリズムをscran・BASiCS・scFeatureFilterなどのパッケージで利用すればOKです。
次元圧縮
遺伝子がたくさんあると人間には理解できないので、次元圧縮を行います。
SingleCellExperimentクラスには、低次元にした跡のデータを保存するためのreducedDimsという専用の置き場があります。
scaterパッケージを使えば、主成分分析(PCA)、t-SNE、UMAPといった代表的な次元圧縮を簡単にできます。
生命現象と結びつけたデータ解析
発現変動遺伝子の探索
複数の群で発現量が変化している遺伝子 (DEG) を知りたいというのはよくあります。
バルクRNA-seqにおけるDEGの探索法として長らく使われてきたedgeRやDESeq2をそのまま使ったり、あるいはzinbwaveのようにたくさんある0を統計学的に減らしてからモデルをフィッティングさせてDEGを探索するアプローチが1つの代表的なグループです。
もう1つのグループは、1細胞解析特有のスパースなデータ専用に開発されたベイズ混合モデルなどをベースにした独自のDEG探索法です。
scDD、BASiCS、scdeなどのパッケージを使って計算できます。
クラスタリング
細胞をクラスタリングしたいというのもよくあります。
グラフベースクラスタリングは、大規模な1細胞RNA‐seqデータ用に作られました。louvainやleidenといったアルゴリズムでクラスタリングを行います。
BiocNeighborsパッケージでは、最も距離が近い細胞と同じクラスターに割り当てるというnearest-neighborと呼ばれる方法でクラスタリングが行われます。
k-means法を1細胞解析データで行ってくれるのがmbkmeansパッケージです。
他にもSC3やclusterExperimentはまた別の方法によってコンセンサス・クラスターを計算してくれるパッケージです。
軌跡分析
シングルセル解析に特化した次元圧縮法の応用である、軌跡解析 (trajectory analysis) や擬似時間推論 (擬似時間推論) では、系統発生的手法を用いて、時間の経過による発生などの軌跡に沿って細胞を順序付けます。
これにより推定された軌跡は、細胞状態間の遷移や分化プロセスなどに使われています。
これを行うためのさまざまなパッケージ (cellTree、LineagePulse、mfa、monocle、slingshot、switchde、TSCAN) があります。
エンリッチメント解析
MSigDB、KEGG、Reactome、GOなどのデータベースのデータを使ってエンリッチメント解析を行うのがEnrichmentBrowserです。
GSEA (gene set enrichment analysis) の1細胞解析版を行うEGSEAやfgseaパッケージもあります。
細胞種の自動割当
SingleRは、細胞種の自動割当を行ってくれる機能があります。ある1細胞について、最も高いスピアマン相関係数を持つレファレンス細胞種と同じという割当を行っています。
正確には、ノイズを抑えるために全ての遺伝子でスピアマン係数を計算しているのではなく、既知の代表的なマーカー遺伝子複数に絞ってスピアマン係数を出しています。
ENCODEやDatabase for Immune Cell Expression(DICE)などから得られたたくさんのレファレンス細胞腫がパッケージに含まれていますが、パッケージに含まれていない細胞種は割当できないという弱点もあります。
進むパイプラインの大衆化
この記事ではさまざまなBioconductorのライブラリーを紹介しましたが、より解析をしやすくするパイプラインの開発も同時並行で進んでいます。そのような状勢について大衆化するシングルセルRNA-seq解析パイプライン 【新しい技術の台頭も】でまとめています。
関連サイト・図書
この記事に関連した内容を紹介しているサイトや本はこちらです。
An introduction to the SingleCellExperiment class
大衆化するシングルセルRNA-seq解析パイプライン 【新しい技術の台頭も】
まとめ
最後に今回の内容をまとめます。
- 1細胞解析データはSingleCellExperimentクラスが基本
- ノイズをきちんと除くことが重要
- たくさんのツールがあるため理解してから使う
今日も【医学・生命科学・合成生物学のポータルサイト】生命医学をハックするをお読みいただきありがとうございました。