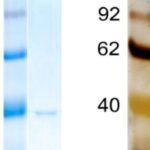
タンパク質の電気泳動には、SDS-PAGEが最もよく使われています。
この記事では、SDS-PAGEのやり方やその後の染色の仕方について、その原理も併せてまとめました。
この記事の内容
SDS-PAGEの原理
DNAの場合は、アガロースを使った電気泳動が一般的ですが、タンパクはアクリルアミドを使います (PAGEはポリアクリアミドゲル電気泳動Poly-Acrylamide Gel Electrophoresis) の頭文字をとったものです。
fa-arrow-circle-right関連記事アガロースゲル電気泳動の方法と原理 【失敗原因の考察も】
SDS-PAGEでは、小さいタンパク質ほど早く移動するため分子量マーカーと比較することでおよその分子量が推定できます。
まずサンプルに SDS と還元剤を加えてボイルします。
SDS は陰極性の界面活性剤で、タンパク質を強力に変性させさせると同時にその疎水性の部分でタンパク質の主鎖と結合します。
2メルカプトエタノール (2ME) などの還元剤は、タンパクのS-S結合を還元して切断します。
その結果タンパク質は完全に変性し、複数のサブユニットからなる場合でも全体がほぼ均一に負電荷を帯びた状態となります。
これをポリアクリルアミドゲル中で電気泳動すると、DNAの電気泳動と同じゲルのふるい効果のため小さいものほど早く流れるのです。
ただし、膜タンパクや糖タンパク、リン酸化タンパク、プロリンの多いタンパクなどでは、SDS結合量が通常のタンパク量と若干違うため、SDS-PAGEでの見かけの分子量が実際の分子量と異なる場合があることに注意が必要です。
タンパク質の泳動度は、ゲルの硬さ (つまり濃度) に依存します。
アクリルアミドの濃度を高くするほど網目が細かくなり、小さいタンパク質を分離するのに適したゲルができます。
一般的には5から15%の濃度を使うことが普通です。
広く行われているSDS-PAGEは、1970年にLaemmliにより報告された手法を踏襲しています。
使用するポリアクリルアミドゲルは、濃縮ゲル(pH6.8)と分離ゲル(pH8.8)の2つのpHが違うゲルを重ねて作ったもので、泳動バッファーとしてTris-Glycine-SDS(pH8.3)を使用します。
このようにすると、まず上にある濃縮ゲルでタンパク質が濃縮され、次に下側の分離ゲルでタンパク質を分子量に応じて分離できます。
より詳しくみてみると、電気泳動開始後、pH6.8の濃縮ゲル中ではタンパク質とバッファー中のTris(25℃で約pKa 8.1)はプラスの電荷を帯びるため陰極側に移動します。
泳動バッファー中のグリシンは濃縮ゲルのpH6.8条件下では両性イオン化するので移動度は小さく、濃縮ゲル中ではタンパク質サンプルは移動度の大きい (下にある) 塩素イオンとグリシンとに挟まれた状態で泳動されます。 各領域の境界では一時的に電圧が大きくなるので、間に挟まっているタンパク質が濃縮されます。
この時には、分離ゲルに近い方 (下側) から順番に塩素イオン、BPB、SDS、タンパク、グリシンの順番に並んでいます。
pH8.8の分離ゲルに到達すると、グリシンはマイナスに荷電するので、タンパク質よりも移動度が大きくなります。グリシンがタンパク質を追い越した後は、タンパク質は分子量の違いにより分離されることになります。
この時には、陽極に近い方 (下側) から順番に塩素イオン、BPB、SDS、グリシン、タンパクの順番に並んでいます。
2つの異なるpHのバッファー中でのイオンの挙動を上手に利用したLaemmli法は、現在でも最も分離性能が良い優秀な方法です。
SDS-PAGEのやり方
SDS-PAGEを行うためには、タンパクを抽出した後、次のような工程が必要になります。
サンプルの調製
電気泳動の開始
それぞれ順番に解説します。
ゲルを作成する
1. ゲル板を組み立てる。泳動板は10 cm四方程度のガラス製のもので、泳動板の間のゲルの厚さが1 mm になるようにスペーサー (ガラスの片面にある'みみ') のついたものを選ぶ.
2. 適当な濃度の分離ゲル用溶液を作り、室温にする。液体の状態のアクリルアミドには神経毒性があるので注意する。
| 6 % | 8 % | 10 % | 12 % | 15 % | |
| 30%アクリルアミド | 2 mL | 2.7 mL | 3.3 mL | 4 mL | 5 mL |
| 1.5M Tris-HCl (pH 8.8) | 2.5 mL | 2.5 mL | 2.5 mL | 2.5 mL | 2.5 mL |
| 10% SDS | 100 ul | 100 ul | 100 ul | 100 ul | 100 ul |
| MilliQ | 5.5 mL | 4.8 mL | 4.2 mL | 3.5 mL | 2.5 mL |
3. 10 mLの分離ゲル用溶液につき、20% APS 30 ulとTEMED 20 ulを加えてよく撹拌した後、ゲル板に上から3 cmぐらいのところまで流し込む。全部は使わず、少し残しておくと、あとでどれくらい固まったのかわかって便利。
4. ピペットマンを使って1ml のイソプロパノールを静かにゲル用溶液の上に重曹 (重しの代わり)。
5. 30分ほどしてイソプロとゲルの境界がはっきりと見えてきたら、ゲルが固まっているので重曹したイソプロを捨てる。
6. 濃縮ゲル用溶液 3 mL (ミニゲルの場合)をとって、20% APS 15 ul, TEMED 5 ulを加え、攪拌し、固まった分離ゲルの上に流し込み、泡が入らないようにコームを差し込む
7. およそ15分ぐらいでゲルが固まる。
サランラップで包み、乾燥しないようにしておけば少なくとも一日は保存可能。
作りたてのゲルでは重合していない古いアクリルアミドが残っている可能性があり、これはシステインと結合するので質量分析で解析する場合は注意が必要。
※ もともと作成済みのゲル (プレキャストゲル) を購入することもできます。割高ですが便利です。
サンプルの調製
1. タンパクサンプル20 ul に、4xサンプルバッファー 6 ulを加える。PCR用のチューブを使うと便利
最終濃度はそれぞれ0.25 M, 8%, 20%。
2. 2-Mercaptoethanol (2ME) を1 ul加える(最終5%)。
3. 88℃で5分 (95℃で3分のプロトコルもある)
4. 軽く遠心して水滴を落とす
※ SDS-PAGE用マーカー (各社から出ている) もサンプルと同じように処理
電気泳動
1. ゲル板からシリコンチューブとコームを外し泳動槽にセット
2. 10xSDS泳動バッファーをMilliQで1xに希釈したバッファーを入れる
最終濃度はそれぞれ250 mM, 1.92 M, 1%。
3. 5 mLのシリンジを使って、ウェルに溜まっている未重合のアクリルアミドと、ゲルの下端 (2枚のガラスの間)の泡をバッファーで吹き飛ばす。
4. 1 ウェルあたり10 ~ 20 L のサンプルをピペットマンで入れる。
5. 電源をつないで100 Vで泳動する。定電流で泳動してもいい。
6. BPB色素がゲルの先端近くになったら泳動を止め、バッファを捨てる。
7. ゲル板を外し、ゲルを取り出す。一部を切り取って上下左右がわかるようにしておくといい。
タンパク質の染色
泳動したタンパクはCoomassie Brilliant Blue (CBB) 染色や銀染色によって検出するのが一般的です。
CBB染色の検出限界は0.1 ~ 0.01 μg程度ですが、簡単にかつ安く染色でき定量性も良好です。
それに対して、銀染色はCBB染色に比べて100倍近く高感度なので、CBBでは検出できない時に有効です。ただし、 CBBに比べタンパク質の種類によって銀発色の発色の違いが大きいという弱点もあります。
CBB 染色
1. プラスチック容器にCBB 染色液を100 mlくらい入れ、そこにゲルを浸し、1 - 2時間浸透
最終濃度はそれぞれ0.25%, 5%, 7.5%
2. 染色液を回収し脱色液で軽く洗う
最終濃度はそれぞれ25%, 7.5%
3. 新しい脱色液で浸透し、青くなったら交換というのを繰り返す (數十分から数時間程度)
4. バックグラウンドが抜けタンパク質のバンドがはっきり見えてきたら、MilliQで10分浸透し、脱色液をMilliQで置き換える。
5. ゲルをスキャンする。ガラス板の周囲にビニールテープを貼り、スキャナーの上面と密着しないようにした状態でスキャナーにおき、ゲルを乗せてスキャン
6. ゲルを濾紙に乗せて、DWでMilliQで濡らしたセロハン紙で覆い、ゲル乾燥機にかける
銀染色
銀染色は固定 (fiation)、増感 (sensitization)、銀染色 (impregnation)、現像 (development) という4つのステップがあります。
それぞれで何を使うかによって多くのプロトコルがありますので、ここでは一例を示します。
1. ゲルを固定液に浸し1時間以上浸透。タンパクを固定するだけでなく、SDSやグリシン、メルカプトエタノールといった、銀染色を妨害する物質を洗い流す意味もある。
最終濃度はそれぞれ25%, 5%
2. MilliQで浸透 (10分 x2)
3. ゲルを増感液に浸し1分浸透
グルタルアルデヒドを使う増感方法もありその方が増感できるが、タンパク質を修飾してしまうので質量分析などができなくなる。チオ硫酸なら銀染色したバンドを切り出して質量分析も可能。
4. MilliQで浸透 (1分 x2)
5. ゲルを銀染色液にひたし、室温で30分浸透
6. MilliQで浸透しながら洗う (30秒 x1)。長く洗いすぎると次の現像の時の発色が弱くなる。
7. ゲルを現像液に浸し、適当な濃さになるまで浸透。強く現像すると68 kDaと54 kDaにバンドが出るが、これはケラチンのコンタミである。
最終濃度はそれぞれ0.036%, 2%。
8. 現像液100 mlにつき酢酸 2 mlを加えて反応を停止。反応停止が不十分だと後で変色する。
9. MilliQで洗う(5分 x3)
SYPRO Ruby染色
SYPRO Rubyという試薬を使って蛍光画像を得ることもできます。感度は銀染色に匹敵。
1. ゲルを固定液に浸し1時間以上浸透
最終濃度はそれぞれ10%, 7.5%
2. ゲルを染色液に浸し1時間以上浸透。
3. ゲルを脱色液に浸透し1時間以上浸透
最終濃度はそれぞれ10%, 7.5%。固定液と同じ。
4. MilliQで攪拌しながら洗う (5分 x2)
5. 蛍光検出装置で画像を取得
SDS-PAGE以外のタンパク電気泳動法
これまで解説してきたSDS-PAGE以外の電気泳動もあります。
Native-PAGE
ウレアを使ったPAGE
Tris-Tricine SDS-PAGE
ここではこれらについて、簡単に紹介します。
非還元 SDS-PAGE
SDS-PAGEでは SDS と還元剤 (2-MEなど) でタンパク質を変性させて泳動しますが、サンプル中のS-S結合を切断したくない場合には還元剤を入れずに移動することがあります。
通常のSDS-PAGEサンプルバッファーから還元剤を除くだけで実施可能です。
注意点として、1枚の同じゲルに還元剤を含むサンプルと含まないサンプルを同時に流した場合、還元剤が拡散して非還元状態のサンプルを還元してしまう可能性があります。
タンパク質の泳動度は分子量に依存しますが、S-S結合でオリゴマーになっている場合はオリゴマーの分子量のところにバンドが出ることになります。
例えばIgGの場合、還元剤を入れていない場合だと、S-S結合で繋がっている2本の重鎖と2本の軽鎖が全て繋がった1本の150 kDaのバンドとして現れます。
Native-PAGE
Native-PAGEは変性剤 (SDS)と還元剤を使わないポリアクリルアミドゲル電気泳動のことです。
SDS-PAGEの系からSDSと還元剤を抜けばいいです。
タンパク質の泳動度は分子量だけでなく電荷に大きく依存するので、適切なバッファーを選び、プラス極とマイナス極のどちらに移動されるかを検討しておかなければいけません。
中性から酸性の等電点を持つタンパク質は多いですが、それらは pH 8.8のバッファー中では負に荷電しているためプラス極側に移動することになります。
等電点が8に近いタンパク質では電荷が少なくなるためあまり泳動しなくなり、さらに等電点が大きいタンパクはマイナス極側に移動することになります。
ウレアを使ったPAGE
Native-PAGEに尿素 (Urea) を加えて移動する方法もあります。
尿素はタンパクを変性させ、変性状態で泳動することになりますが、Native-PAGEと同じくタンパク質自身の電荷で移動するので移動度はタンパク質の電荷を反映します。
リン酸化状態の違いを検出したい時、分子量の近い複数のタンパク質を分離したい場合に使われています。
Tris-Tricine SDS-PAGE
SDS-PAGEで検出できる最も小さいタンパクは分子量12000程度です。
さらに低分子のタンパク質を検出したい場合には、グリシンの代わりにTricine を使った SDS-PAGEを使うといいです。
分子量3000から1万くらいのタンパク質を分離できます。
これよりも小さい場合、通常の染色操作を行う時にゲルから抜けてしまう可能性があり、検出は困難かもしれません。
関連サイト・図書
この記事に関連した内容を紹介しているサイトや本はこちらです。
まとめ
最後に今回の内容をまとめます。
- タンパクの泳動はSDS-PAGEが基本
- 泳動後はCBBや銀染色などを行う
- SDS-PAGE以外の泳動方法もある
今日も【医学・生命科学・合成生物学のポータルサイト】生命医学をハックするをお読みいただきありがとうございました。