ChIP-on-chipは、生命科学研究手法の1つです。
クロマチン免疫沈降(chromatin Immunoprecipitation:ChIP)とマイクロアレイを組合わせることで、網羅的に転写因子結合領域やクロマチン修飾を探索することができます。
今でこそ次世代シークエンサーと組み合わせたChIP-seqがよくやられていますが、次世代シークエンサーが出る前のマイクロアレイ全盛期にはよくやられていた方法であり、先人たちが取得したデータが公共データとして誰でも利用可能になっています。
そこで、公共データを使ったChIP-on-chipの再解析の例として、この記事ではデータのダウンロードの方法から統計ソフトRを使った具体的なコードを紹介します。
この記事の内容
データをArrayExpressからダウンロードする
まずはデータをダウンロードしましょう。
国際的に有名なデータアーカイブとしてGEOとArrayExpressが知られていますが、ここではArrayExpressにアクセスし、E-GEOD-31301を検索してみます。

Saccharomyces cerevisiae (出芽酵母) を対象とした、Isw1-3xFLAGを発現させてFLAGでChIPし、lsw1の結合部位を調べた研究であることが分かります。
ArrayExpressには生データだけでなくいろいろ正規化したProcessed dataがあるので、今回はこちらを使ってみます。

「E-GEOD-31301.processed.1.zip」データをダウンロードし、解凍し、Rのワーキングディレクトリーに置いてください。
その下にあるArray designは、マイクロアレイのどの場所のプローブがゲノム上のどの部位に相当するのかをまとめたもので、これも必要です。A-GEOD-13972.adf.txtファイルを同じくRのワーキングディレクトリにおいてください。
これで下準備は終了です。
データをRに読み込む
データはいずれもテキストファイルなので、特別なパッケージを使わずにRに読み込むことができます。
例えば「GSM775491_sample_table.txt」というファイルと「A-GEOD-13972.adf.txt」を読み込んでみます。
Rにテキストファイルを読み込むには、read.delim関数を使うのが簡単です。
my.dat <-read.delim("GSM775491_sample_table.txt",row.names=1)
adf <- read.delim('A-GEOD-13972.adf.txt',skip=15, row.names=1)
names(adf) <- c("Coordinate","Sequence")どんな風に読み込めたのか確認してみましょう。まずはGSM775491_sample_table.txtについては、
head(my.dat)このように、各プローブでの検出値が出てきます。
VALUE
4 -0.145817615
5 -0.006284506
6 0.116073348
7 0.784466893
8 -0.143302852
9 -0.102563881
IP/inputの値をlog2変換したものですので、数値が正であればFLAG抗体でIPした時により濃縮されていて、逆に負であればIPすると減ってしまうことを意味しています。
同様にA-GEOD-13972.adf.txtは
head(adf)Coordinate
1
2
3
4 chr4:000179775-000179834
5 chr3:000071144-000071192
6 chr3:000239622-000239681
Sequence
1
2
3
4 ATTCCCCAATTAGGATACTTAGTAACCATTAGCGTCTTAATGGAACCTTTATTGGATGGC
5 CAAGTTCCTACCTGGGCAATAACTCCAATTCAAATTCGAGTTATGGGGG
6 AACTTCTGGTCTAGATCAACTAATGGATGCTATTCAATACACAGAAACAGACGAATATGG
このようになります。Cooridnateはゲノムの場所、Sequenceはプローブの実際の配列です。
1-3列目は空白ですが、これは蛍光補正などに使うコントロールプローブであることを意味しています。
プロセス後のデータを使う限りは、これらのコントロールプローブには用がないのでここでは割愛します。
ChIP-on-chipデータの前処理
coorinate列にはchr3:000071144-000071192のように染色体番号:開始位置-終了位置という形式でプローブ情報が書かれています。
これらは単なる文字情報ですので、R組み込みの関数を使ってそれぞれの情報を抽出していきます。
また染色体番号がわからないプローブがいくつかあるので、それについては除いておきます。
adf$chr <- apply(matrix(adf$Coordinate, ncol=1),1,function(coord){strsplit(coord,':')[[1]][1]})
adf$pos <- apply(matrix(adf$Coordinate, ncol=1),1,function(coord){strsplit(coord,':')[[1]][2]})
adf$start <- apply(matrix(adf$pos, ncol=1),1,function(coord){strsplit(coord,'-')[[1]][1]})
adf$end <- apply(matrix(adf$pos, ncol=1),1,function(coord){strsplit(coord,'-')[[1]][2]})
adf <- adf[!is.na(adf$chr),] Coordinate列はRに文字として認識されていて、そこから情報を抽出しても文字のままになりますが、解析をする上でそれは困るので、染色体番号はfactor型、染色体の開始位置と終了位置は数値型に変換しておきます。
adf$start <- as.numeric(adf$start)
adf$end <- as.numeric(adf$end)
adf$chr <- as.factor(adf$chr)
head(adf)これを実行すると、もともとのCoorinate, Sequence列だけでなく、その後ろにchr (染色体番号), pos (位置), start (開始部位), end (終了部位)を抜き出せていることが分かります。
Coordinate
4 chr4:000179775-000179834
5 chr3:000071144-000071192
6 chr3:000239622-000239681
7 chr14:000285414-000285473
8 chr7:000835520-000835578
9 chr10:000492976-000493035
Sequence chr
4 ATTCCCCAATTAGGATACTTAGTAACCATTAGCGTCTTAATGGAACCTTTATTGGATGGC chr4
5 CAAGTTCCTACCTGGGCAATAACTCCAATTCAAATTCGAGTTATGGGGG chr3
6 AACTTCTGGTCTAGATCAACTAATGGATGCTATTCAATACACAGAAACAGACGAATATGG chr3
7 CCATTAGTTAAACTTTTGGAAGTTGCAGAATATAAAACTAAAAAAGAAGCTTGTTGGGCT chr14
8 AGGATACTCTCATCTTCAAAAACAATATTGAATCCCGCTATTCTGCCAGGTAAATCCTC chr7
9 AAATCCATATCATGCACTTGCTTGCAAAGAACGTTGACACAATCTTTTAGAGAGTCAGTT chr10
pos start end
4 000179775-000179834 179775 179834
5 000071144-000071192 71144 71192
6 000239622-000239681 239622 239681
7 000285414-000285473 285414 285473
8 000835520-000835578 835520 835578
9 000492976-000493035 492976 493035
3列目(chr), 5列目 (start), 6列目 (end) の情報を、蛍光強度の情報を持つmy.dat変数に列方向に連結します。
adf[,c(3,5,6)]
my.dat <- cbind(my.dat, adf[,c(3,5,6)])
head (my.dat)VALUE chr start end
4 -0.145817615 chr4 179775 179834
5 -0.006284506 chr3 71144 71192
6 0.116073348 chr3 239622 239681
7 0.784466893 chr14 285414 285473
これでそれぞれのプローブの蛍光強度と、そのゲノム上の位置が一つになった表を得ることができました。
例えば、染色体ごとのプローブ数については
table(my.dat$chr)chr1 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9
1015 3685 3435 5284 4715 3929 5497 4760 4114 1528 7657 2852 1320 5475 2760 2171
chrM
450
のように調べることができます。
ChIP-on-chipデータの絞り込み (デモ目的)
もちろん全ての染色体について調べてもいいのですが、この記事ではデモ目的として最もプローブが多く設計されている染色体4に絞って解析方法をみていきます。
絞るためには、Rのスライスを使えばいいですね。
my.dat.c4 <- my.dat[my.dat$chr=='chr4',]さらに開始位置 (start) で並び替えます。
my.dat.c4 <- my.dat.c4[order(my.dat.c4$start),]これを図示してみましょう。
w.start <- min(my.dat.c4$start)
w.end <- max(my.dat.c4$end)
plot(my.dat.c4$VALUE ~ my.dat.c4$start, t='l',xlim=c(w.start,w.end),main="ChIP-Chip peaks\non Chromosome 4",xlab="Position",ylab="log2 IP/Input")
abline(h=0,col="blue",lty=3)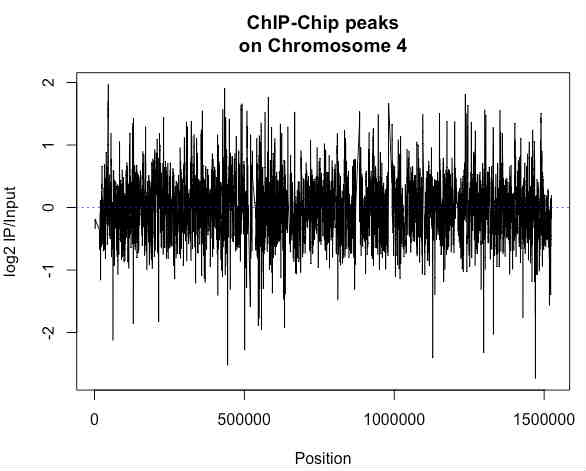
染色体4番全体でみてみると、あちこちにピークがあるので、ここでは500,000から512,000までの12000塩基の領域に絞ってみます。
w.start <- 500000
w.end <- 512000
plot(my.dat.c4$VALUE ~ my.dat.c4$start,t='l',xlim=c(w.start,w.end),main="ChIP-Chip peaks\non Chromosome 4",xlab="Position",ylab="log2 IP/Input")
abline(h=0,col="blue",lty=3)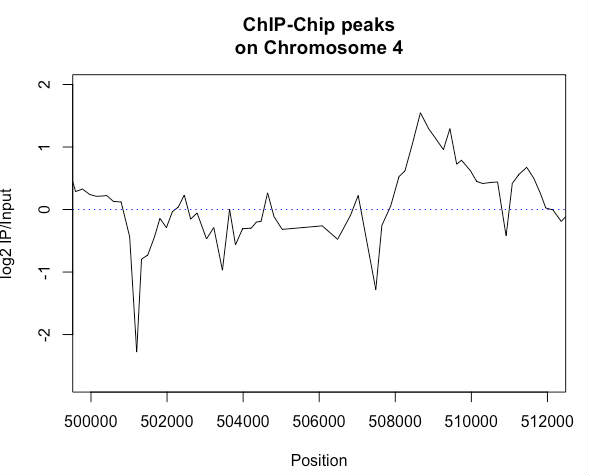
次にやるのはこのデータをもとにしたピーク検出です。
ChIP-on-chipデータのピーク検出
領域を絞ることで、ピークがより鮮明になりました。特に、501,100にある負のピークはかなりシャープであり、本当のピークのように見えます。
しかしこれは間違いで、1つのプローブ情報だけでは偽物のピークが出てしまう可能性があります。統計学で多重補正が必要なのと同じ理屈です。
ピークの検出には、近隣のプローブ情報も加味したスライディングウインドウというアプローチを行います。
これはある一定の範囲 (ウインドウ) の情報を平均することで、「たまたま」出てしまったシグナルの影響を取り除くことができる一方で、「本当の」結合部位は近隣の複数のプローブでも検出できることから平均してもシグナルのまま残るということです。
例えば、ウインドウ幅を1000塩基にして平均し、この領域全体を少しずつウインドウをずらしながら計算して先ほどの図の上に赤で描いてみます。
w.size <- 1000 # windowサイズを1000とする
w.pos <- rep(NA,(w.end-w.start)/w.size) # 位置を格納するベクトルをNAで定義しておく
w.smooth <- w.pos
i <- 1
for(pos in seq(w.start,w.end+w.size,w.size)){
vals <- my.dat.c4$VALUE[my.dat.c4$start>pos & my.dat.c4$start<(pos+w.size)] # その範囲に入るシグナルを全部格納しておく
w.pos[i] <- pos
w.smooth[i] <- mean(vals) # 複数のシグナルの平均値で代用
i <- i+1
}
plot(my.dat.c4$VALUE ~ my.dat.c4$start,t='l',xlim=c(w.start,w.end),main="ChIP-Chip peaks\non Chromosome 4",xlab="Position",ylab="log2 IP/Input")
abline(h=0,col="blue",lty=3)
lines(w.pos,w.smooth,col="red",lty=2)
すると、左側にあった負のピークは消え、一方で508,000前後の正のピークはそのまま残っていて本当のシグナルだと考えられます。
実際、この部位は実験的にlsw1タンパクが結合することが確かめられている場所になります。
このようにChIP-on-chipのデータは特別なプラグインなしで公共データからダウンロードして解析することが可能です。
興味があるタンパクについてのデータがあればぜひやってみてください。
関連サイト・図書
この記事に関連した内容を紹介しているサイトや本はこちらです。
まとめ
最後に今回の内容をまとめます。
- ChIP-on-chipデータはGEOやArrayExpressからダウンロードできる
- スライディングウインドウをすることに注意
- ChIP-on-chipのデータは特別なプラグインなしで解析できる
今日も【医学・生命科学・合成生物学のポータルサイト】生命医学をハックするをお読みいただきありがとうございました。